
Full Text View
Volume 19 Issue 10 (October 2009)
GSA Today
![]()
Article, pp. 4-10 | Abstract | PDF (779KB)
Did natural reactors form as a consequence of the emergence of oxygenic photosynthesis during the Archean?
| Table of Contents |
|---|
| Search GoogleScholar for
Search GSA Today |
The advent of oxygenic photosynthesis changed Earths surface environment in numerous ways, perhaps most notably by making possible the evolution of large and complex life-forms. Current models suggest that organisms that can perform oxygenic photosynthesis first took hold in isolated marine and freshwater basins, producing local oxygen oases. Here we present calculations that suggest that uranium deposits could have formed at the margins of these basins due to the strong local reduction-oxidation gradients. Because of the high abundance of 235U at this time, these uranium deposits could have formed widespread, near-surface, critical natural fission reactors. These natural reactors would have represented point sources of heat, ionizing radiation, and free radicals. Additionally, they would have far-field effects through the production of mobile short- and long-lived radioactive daughter isotopes and toxic byproducts. It is possible that these fission products provided a negative feedback, helping to limit the proliferation of the cyanobacteria in the Archean environment. Secular decreases in the abundance of 235U in turn decreased the probability of such deposits forming critical fission reactors during the early Proterozoic.
Manuscript received 14 January 2009; accepted 30 July 2009.
doi: 10.1130/GSATG41A.1
*E-mails: ,
OXYGENATION OF EARTHS ATMOSPHERE
An oxygenated atmosphere is a prerequisite for the evolution of complex life. On Earth, atmospheric oxygen is produced through oxygenic photosynthesis. It is widely, although not unanimously (e.g., Ohmoto et al., 2006), accepted that oxygen levels in Earths atmosphere were very low throughout the first ~2 Ga of Earths history (Fig. 1). Evidence from paleosols for soil development under reducing conditions and the occurrence of clastic sediments containing minerals that are highly soluble under oxic conditions, such as pyrite and uraninite, suggest low atmospheric oxygen before ca. 2.3 Ga (e.g., Rye and Holland, 1998). Evidence for low atmospheric oxygen before this time also comes from the occurrence of Δ33S anomalies in sediments older than ca. 2.4 Ga that are generally believed to originate through photochemical reactions in an essentially O2-free atmosphere (e.g., Farquhar et al., 2000; Bekker et al., 2004; Domagal-Goldman et al., 2008). Despite this, there is evidence that local oxygenated oases existed prior to this time. For example, the Pb-isotopic composition of some >3.7 Ga metasediments suggests that their protoliths had elevated primary U/Th ratios (Rosing and Frei, 2004). This is interpreted as indicating that oxidized, and thus mobile, U was locally reduced and hence accumulated in the sediment. Additionally, local enrichment of Re and Mo, two other redox sensitive elements, in 2.5 Ga sediments suggests local oxygen oases at this time (Anbar et al., 2007).
|
Estimated evolution of atmospheric oxygen content over Earths history (e.g., Rye and Holland, 1998; Catling and Claire, 2005; Canfield, 2005; Holland, 2006). The likely timing of the evolution of oxygenic photosynthesis, indicated by bar A (Schopf, 1993) to B (Brocks et al., 1999) significantly precedes the large increase in atmospheric oxygen indicated by the end of mass-independent S-isotope fractionation (C; Domagal-Goldman et al., 2008). During this time, local oxygen oases may have existed. |
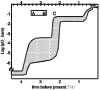 Figure 1
Figure 1The suggestion that local oxygen oases existed prior to the widespread oxygenation of the atmosphere is consistent with evidence that oxygenic photosynthesis may have evolved very early in Earths history (Fig. 1; see also Rasmussen et al., 2008). Fossils resembling cyanobacteria have been documented in ca. 3.5 Ga cherts (Schopf, 1993), and 3.2 Ga organic-rich, but pyrite-poor, sediments are difficult to explain unless oxygenic photosynthesis had evolved by this time (Buick, 2008). Likewise, C-isotope evidence for organisms using RuBisCO−1 (the enzyme that catalyzes CO2 fixation during oxygenic photosynthesis), alongside other geochemical evidence, suggests that oxygenic photosynthesis had evolved by 2.9 Ga (Nisbet et al., 2007).
The existence of oxygen oases prior to widespread oxygenation of the atmosphere is consistent with ideas about the evolutionary transition from anoxygenic to oxygenic photosynthesis, which is apparently paradoxical as it requires an already somewhat oxidizing environment. Oxidizing environmental conditions are necessary to select for the biochemical machinery capable of extracting electrons from water and to develop the cellular defenses to cope with acute oxygen toxicity (McKay and Hartman, 1991; Blankenship and Hartman, 1998). A model for the evolution of oxygenic photosynthesis that can address this paradox involves an origin in isolated marine basins or terrestrial lakes where H2O2, produced photolytically in the Archean atmosphere (Kasting et al., 1985), could act as an oxidant (McKay and Hartman, 1991). H2O2 rained out of the atmosphere would have oxidized the common anoxygenic electron donors (Fe2+ and H2S) in the photic zone. Decreasing availability of these electron donors would increase the selection pressure to use H2O as a reductant. The presence of H2O2 in surface waters facilitates the production of more aggressive free radicals like OH· through the Fenton and similar reactions (for example):
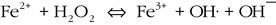 (1).
(1).
The presence of such reactive oxygen species requires that ancient phototrophs adapted biochemical strategies to cope with oxidative stress even before the first molecule of biologically produced O2 was released into the environment. This model of H2O2 induced oxidative stress prior to the emergence of oxygenic photosynthesis provides the selective pressure to extract electrons from water and equips the predecessors of oxygenic photoautotrophs with the cellular machinery to cope with reactive free O2 (Blankenship and Hartman, 1998). If correct, biological O2 production first took hold in isolated marine and freshwater environments and must have established oxidizing microenvironments bounded by strong local reduction-oxidation gradients.
DELAYED OXYGENATION OF THE ATMOSPHERE
Evidence for the evolution of oxygenic photosynthesis and the existence of oxygen oases prior to global oxygenation of the atmosphere ca. 2.4 Ga raises the question of what delayed the proliferation of oxygenic photoautotrophs and oxygenation of the atmosphere. Numerous models have been proposed. One class of model suggests that the sinks for photosynthetically produced oxygen decreased ca. 2.4 Ga. This requires that the rate of volcanic and/or metamorphic release of reduced gases, and/or the oxidation state of these gases, changed at this time (Kasting et al., 1993; Kump et al., 2001; Holland, 2002; Kump and Barley, 2007). However, there is little evidence for a change in the oxidation state of Earths mantle over geological time (Canil, 1997; Li and Lee, 2004), and the evidence for changing rates of volcanism is ambiguous.
A second model suggests that if the Archean atmosphere had been methane-rich, significant H could have been lost into space, leading to irreversible oxidation of Earths surface (Catling et al., 2001). There is uncertainty over whether the Archean atmosphere was methane-rich (e.g., it is unclear how, under a CH4-rich atmosphere, local oxidative environments could have existed as is apparently required to provide the selection pressure allowing oxygenic photoautotrophs to evolve). Additionally, hydrogen loss depends on the temperature at the exobase, the height in the atmosphere above which there are the negligible collisions between molecules and escape into space is possible, and it is currently unclear whether this temperature was high enough in the Archean to allow H-escape rates much greater than modern values (Tian et al., 2005).
A further set of models suggests that Earths atmosphere may have two stable states—oxygen-poor and oxygen-rich. This may be because of nonlinear changes in the lifetime of oxygen in the atmosphere as oxygen and, hence, ozone concentrations increase and provide a shield from ultraviolet radiation (Goldblatt et al., 2006). Alternatively, low O2 and CO2 concentrations may be stable in a CH4-rich atmosphere and vice versa. In this scenario, increased atmospheric O2 levels under a CH4-rich atmosphere would have been inhibited by low CO2 levels limiting photosynthetic O2 production and because reaction of O2 with CH4 would decrease the greenhouse effect, potentially inducing glaciation (Nisbet et al., 2007).
Finally, it has been proposed that links between the oxygen and nitrogen cycles were responsible for the delay between evolution of oxygenic photosynthesis and appearance of an oxygen-rich atmosphere (Falkowski and Godfrey, 2008). In this model, oxygen production by early oxygenic photoautotrophs led to oxidation of oceanic ammonium to nitrate followed by denitrification of the oceans. Because fixed nitrogen is an essential nutrient, a decrease in its availability in the oceans would have acted as a negative feedback on the production of oxygen.
Based on consideration of the geological setting where oxygenic photosynthesis likely emerged, we suggest an additional factor that would have had a negative impact on the local environment of the earliest photoautotrophs. Natural fission reactors may have formed at the margins of local oxygen oases, as illustrated in Figure 2 and explored next.
|
Cartoon showing a possible mechanism by which oxygenic photosynthesis could lead to formation of natural fission reactors. Uraninite weathered out of igneous and metamorphic rocks is transported to isolated basins and deposited in shallow water environments, providing a ready source of U as soon as the waters become oxidizing. Photolytically produced H2O2 rains out of the atmosphere and oxidizes the uppermost water column, reducing the concentration of electron donors required by anoxygenic photosynthesizers such as H2S and Fe2+. This provides the selective pressure required for the emergence of oxygenic photosynthesis due to the abundance of H2O as an alternative electron donor. Once oxygenic photosynthesis evolves, this produces local enrichment in oxygen in the surface water. This would mobilize uranium into solution (see Figs. 3 and 4), which would be redeposited at the margins of the oxygen oasis. These uranium deposits would have the potential to form natural reactors due to the high concentration of 235U in the Archean. |
 Figure 2
Figure 2THE GEOLOGICAL SETTING OF EARLY OXYGEN OASES AND URANIUM MOBILITY
Under an anoxic Hadean and Archean atmosphere, uraninite (UO2) weathered out of igneous and metamorphic parent rocks was relatively insoluble and was transported to depocenters. Due to its much greater density (~10 gcc−1) than average sedimentary detritus, uraninite will have tended to be hydrodynamically separated from silicate minerals during transport and deposition; this is the proposed origin of many Archean U deposits. Archean placer uraninite deposits can be highly concentrated, locally containing 30.5 wt% uraninite, or they can be of lower grade but extremely widespread, extending many kilometers laterally (Kimberley, 1978; Theis, 1978). Very early in Earths history (~4.3 Gyr ago) the concentration of 235U may have been high enough for these detrital uraninite deposits to go critical, forming natural fission reactors (Adam, 2007). It is unlikely however that detrital uraninite deposits were sufficiently concentrated for natural reactors to form this way within the mid- or late-Archean, largely due to the secular decrease in the activity of 235U.
Because of the insolubility of uraninite during transport in the Archean, the clastic sediments surrounding the isolated basins in which oxygenic photosynthesis evolved must have contained uraninite either dispersed within the sediments or locally concentrated with other heavy minerals in placer deposits. Photosynthetically produced oxygen would lead to local oxygenation of surface waters. Uraninite becomes highly soluble in the presence of trace oxygen partial pressures (Fig. 3) meaning that uraninite dissolution would occur, thus mobilizing U from the sediments into solution. The rate of uraninite dissolution depends on the pH, bicarbonate, and oxygen concentration of the solution (Ono, 2001). The lifetime (tlifetime) of a spherical uraninite grain can be determined from (Lasaga, 1998):
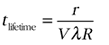 (2),
(2),
where r = grain radius, λ = surface roughness (assumed = 10), V = molar volume (uraninite = 27 cm3mol−1), and R is the dissolution rate (mol m−2 sec−1) determined experimentally as a function of pH, bicarbonate, and pO2 (Ono, 2001).
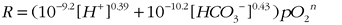 (3),
(3),
where the proton and bicarbonate concentrations are in mol L−1 and pO2 is relative to present atmospheric level (PAL). The order of the reaction with respect to oxygen (n) is uncertain (between 0 and 1), but the experimental study by Ono (2001) suggests an order of n = 0.27. Figure 4 shows that an idealized 200-µm-diameter uraninite grain would completely dissolve in <10 ka at local partial pressures of O2 ~2% of PAL irrespective of atmospheric CO2 concentrations (i.e., uraninite dissolution is expected to have been rapid in the surface water of oxygen oases).
|
The stability fields of different U-species (red lines), shown as a function of pH and ƒO2 calculated assuming T = 70 °C, ƒCO2 = 5 atm, and modern seawater concentrations for the major ions. In modeling this phase diagram, uranium activity of 10 nmol/L and Fe activity of 50 μmol/L were used to reflect increased Fe solubility in the anoxic Archean ocean. The stability fields of Fe-species is overlain (separated by blue lines), with the stability fields of the minerals hematite, siderite, and Mg-ferrite shaded in blue. Note the predicted mobility of U with increasing ƒO2 while Fe, a potential neutron absorber, remains in the solid phase for pH >5.8. |
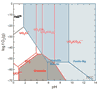 Figure 3
Figure 3|
Lifetime of a spherical uraninite grain (200 μm diameter) for different partial pressures of oxygen relative to present atmospheric oxygen (PAL) for various solution pH. Calculated according to Ono (2001) with ALK = 1.3 mM and an oxygen dependence of n = 0.27; pCO2 is 1 times PAL for pH = 8.3 and roughly 2200 times PAL for pH = 5.0. |
 Figure 4
Figure 4|
The critical mass of uranium needed to produce a natural fission reactor has changed over Earths history because the proportion of uranium that is fissile 235U has decreased dramatically. This is shown by the right-hand axis, and exponentially decaying thin dashed line, that illustrate the relative proportion of U that was fissogenic 235U as a function of time. The mass of U required for a natural reactor to go critical is shown as a function of age for different concentrations of uraninite in the reactor core (15 modal percentage shown with solid lines) assuming a reactor comprised of quartz, uraninite, and water. The thick dashed line shows the increase in the mass of U required at any given time due to adding 10% ilmenite to the most uraninite-rich reactor core (5%) because the Fe and Ti act as neutron poisons. Although the exact mass of uranium required for a natural fission reactor depends on poorly constrained compositional and geometric factors, it is clear that the mass of uranium required during much of the Archean was very small (<1000 kg, or ~0.1 m3) and then increased nearly exponentially as the abundance of 235U decreased at some time around the Archean-Proterozoic boundary. Comparison of the calculated mass of uranium required with the mass observed in the Oklo reactors provides some ground-truth to the models. Input parameters for the calculations were ε = 1.004 following Adam (2007) (the exact value plays a secondary role in the calculations); for r, we used the relationship between uranium concentration and resonance escape probability derived by Adam (2007; his Fig. 2); see text for further details. |
 Figure 5
Figure 5Fluid flow out of the oxygen oases would act to trap the mobilized U at the limits of the oxygenated zone due to the insolubility of U4+, potentially generating significant U accumulations. This mechanism of uranium concentration through the reduction of U6+ to U4+ at redox boundaries is the principal way high-grade Proterozoic and Phanerozoic U deposits formed, demonstrating the efficiency of this process in concentrating U. The dissolution of detrital uraninite, and its subsequent reprecipitation upon reduction, will have purified the uraninite, removing elements that act as potent neutron poisons (e.g., rare earth elements [REEs]) within a natural reactor.
NATURAL REACTORS—GEOLOGICAL INSIGHTS FROM OKLO AND BANGOMBÉ
In the Archean, the abundance of the fissile 235U isotope was much higher than today and thus relatively small uranium deposits could potentially have gone critical (Fig. 5). The potential for U deposits to have formed natural reactors early in Earths history was recognized by Kuroda back in 1956. Thirty years after Enrico Fermi built a critical fission reactor in a squash court in Chicago, it was discovered that nature had achieved this phenomenon ~2 Ga earlier. This realization came from the observation that uranium deposits at Oklo and Bangombé, separated by ~30 km but both within the Franceville intracratonic basin in Gabon, are depleted in 235U relative to 238U and enriched in numerous isotopes produced by fission reactions (e.g., Gauthier-Lafaye et al., 1996; Hidaka et al., 1999).
These uranium deposits, seventeen of which have been shown to have acted as natural fission reactors, are hosted in relatively unmetamorphosed fluviatile and deltaic sequences of conglomerates and sandstones, some of which were deposited in tidally influenced environments ca. 2150 Ga (Gauthier-Lafeye and Weber, 2003). Some of the conglomerates contain relatively high concentrations of U (and Th) in detrital phases. This suggests that concentrations of heavy minerals, potentially including uraninite, in these units may have been the source of uranium for the natural reactors (Gauthier-Lafeye and Weber, 2003). Uranium was mobilized from these conglomerates by post-deposition oxidized basinal fluids and subsequently reduced, and hence deposited, upon interaction with organic-rich reduced fluids in structurally controlled traps (Gauthier-Lafeye and Weber, 2003).
The cores of the natural reactors vary in size: One of the largest and best studied is 12 m × 18 m × 0.20.5 m (“reactor 2”); a smaller reactor at Bangombé is 5 m × 1 m × a few centimeters (Gauthier-Lafeye and Weber, 2003). The cores of the reactors currently have high uranium concentrations (20%–60%), but self-sustaining fission is thought to have initiated when the uranium concentration reached ~10%, and subsequent uranium concentration occurred through the dissolution and removal of quartz in hydrothermal fluids driven by the heat from the fission reactions (Gauthier-Lafeye and Weber, 2003).
The lifetime and energy budget of the Oklo reactors have been estimated in a number of ways. Isotopic and modeling studies suggest that the reactors operated for 2 × 104 to 2 × 105 years (Hidaka and Holliger, 1998) and produced ~5 × 1017 J (Gauthier-Lafaye et al., 1996; Petrov et al., 2006). This is equivalent to a steady-state power output of ~100 kW, although it is unlikely heat output was steady. Instead, high energy production likely led to the pore water, which acted as a moderator slowing neutrons, being driven out of the system, thereby shutting down the reactors until they cooled and water could replenish the pores (Meshik et al., 2004). As well as producing energy and fissogenic isotopes, many with short half-lives, radiolysis produced O2, H2, and H2O2, which are all observed in fluid inclusions within the rocks surrounding the reactor cores (Mathieu et al., 2001; Gauthier-Lafeye and Weber, 2003). The exact reasons the reactors stopped operating are unknown, but it is known that fissogenic light rare earth elements (LREEs), which act as neutron poisons, were retained in uraninite, which must have had a negative effect on fission reactions (Gauthier-Lafaye et al., 1996).
The preservation, and near surface exposure, of any natural reactors is remarkable and suggests that this was not an isolated incident. Archean uranium deposits formed near the surface would have a low preservation potential in the rock record. This is illustrated by the age distribution of epithermal Ag-Au deposits. These have an average formation depth of ~0.5 km and a modal age of only 3 Ma (Wilkinson and Kesler, 2007). Modeling their age distribution suggests that <1% of all mineral deposits formed at this depth over the Phanerozoic are exposed at the surface today (Wilkinson and Kesler, 2007). Considering the smaller portion of Earths surface covered with Archean and early Proterozoic rocks, and the much greater time since formation for erosion to destroy the deposits, the preservation of the remains of any near-surface natural fission reactor suggests that these might have been relatively common in the late Archean and early Proterozoic.
NATURAL REACTORS—THEORETICAL CONSIDERATIONS
A critical fission reactor requires that the number of fission-inducing neutrons emitted per fission is ≥1. The mass of uranium (235U and 238U combined, ignoring minor isotopes) required for a natural critical fission reactor has changed over Earths history as the amount of fissile 235U has decreased dramatically (235U has a half-life of ~707 Ma vs. ~4470 Ma for 238U; Fig. 5). The mass of uranium required for criticality can be calculated given estimates of the shape and composition of uranium deposits, but to extrapolate back through geological time, the changing abundances of 235U and 238U due to radioactive decay (to form Pb) must be accounted for. We followed the general approach described by Adam (2007) and the DOE “nuclear physics and reactor theory” handbook (1993), an approach that goes back to Fermi (1947), to determine the mass of uranium required to form a natural reactor. Because neither the composition nor shape of possible deposits are known, we emphasize the relative change in mass of uranium required over time rather than the absolute mass of uranium required at any given time.
Assuming a lens-like uranium deposit, we modeled the reactor as a cylinder. For this geometry, the number of fission-inducing neutrons released per fission (k) can be determined from the so-called four-factor equation (numerator in Eq. 4) corrected for leakage of neutrons from the margins of the cylinder (denominator in Eq. 4). The first two factors in the numerator of Equation 4 give the efficiency with which neutrons are thermalized (slowed), and the latter two terms reflect the efficiency of thermal neutron absorption in inducing further fission within the reactor:
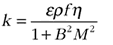 (4)
(4)
and
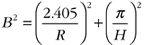 (5),
(5),
where ε = the ratio of the total number of induced fissions to the number of fissions induced by thermal (i.e., slow) neutrons (~1 for an efficient reactor moderator); ρ = the probability of a fast neutron not being absorbed by a 238U nucleus prior to thermalization (i.e., during slowing); ƒ = the ratio of the number of thermal neutrons absorbed by uranium to the number of thermal neutrons absorbed within the reactor, which depends on the bulk composition; η = the ratio of the number of fast neutrons produced by fission to the number of thermal neutrons absorbed by uranium; B = geometrical term dependent on reactor shape—here a cylinder of height (H) and radius (R); M = neutron migration length. Further details are given in Adam (2007) and DOE (1993).
Figure 5 shows that the mass of U required to produce a natural reactor during much of the Archean was small (<1000 kg, or ~0.1 m3) and then increased nearly exponentially as the abundance of 235U decreased at some time around the Archean-Proterozoic boundary. The small masses of uranium needed to form a natural fission reaction during the Archean supports the suggestion that they were fairly widespread. The greatest uncertainty in calculating the mass of uranium required to form a natural reactor back through time comes from the lack of constraint on the bulk composition of uranium deposits over geological time (both mineralogy and water content). In computing the curves shown in Figure 5, we assumed a very simple system composed of quartz + water + uraninite ± ilmenite. High concentrations of neutron poisons, such as would be the case for a system containing B- and REE-rich minerals, would require more uranium for a given age. This is illustrated with the example containing 10% ilmenite (both Fe and Ti have neutron absorption cross sections approximately an order of magnitude greater than the other major elements in Earths crust [Si, Al, Ca, Mg] but still orders of magnitude smaller than poisons such as B).
WERE NATURAL FISSION REACTORS IMPORTANT FOR THE EARTH SYSTEM?
The preceding discussion suggests that natural fission reactors may have been common at the margins of the oxygen oases produced by early oxygenic photoautotrophs. During this time, uraninite occurred as a detrital phase, providing a ready source of U to surface waters as soon as they became oxidized. A key question is whether the natural reactors would have negatively impacted the proliferation of these photoautotrophs, potentially helping to explain the delayed oxygenation of Earths atmosphere (Fig. 1).
Natural reactors act as point sources of heat, ionizing radiation, short- and long-lived radioactive daughter isotopes, and toxic byproducts. Reactors can thus have near- and far-field negative effects on oxygenic photosynthesizers through desiccation, thermal sterilization, ionizing radiation, and toxicity. Temperature reconstructions of the Archean suggest that early oxygenic photosynthetic organisms were likely living near, or at, their maximum allowable temperature (Lowe and Tice, 2007; Gaucher et al., 2008). Natural reactors in near-surface sediments in shallow water would act to raise local water temperatures. However, given that critical natural reactors were likely moderated principally by water, their power output is limited by the temperature and pressure of near-surface water bodies and would have had typical power outputs in the kilowatt range (Draganic et al., 1983), which is insignificant on the scale of a lake or isolated marine system. Ionizing radiation doses in the krad h−1 (~10s Gy h−1) range, significantly higher than natural background levels of ~5 × 10−7 Gy h−1, would be produced in the core of small natural reactors. The co-location of reactors with oxygen oases would increase the rates of radiogenic DNA damage to early oxygenic photoautotrophs through the strong modifying influence of molecular oxygen on free radical production (Karam and Leslie, 1999; Karam et al., 2001). While direct radiation from the core would be rapidly absorbed with distance, part of this energy would generate potent free-radicals in local oxygenated waters. Perhaps the most significant feedback on early oxygenic photoautotrophs would be migration of short- and long-lived fissiogenic radionuclides from reactor cores to the environment. For a reactor operating at 1 kW over 105–106 years, ~14% of product radionuclides have half-lives greater than a year (Draganic et al., 1983). Among these product atoms are elements that would likely be fixed into local inorganic substrates or ultimately incorporated into organic matter (e.g., 90Sr, 137Cs, 135Cs, and 129I) where they would serve as specific radiation sources for centuries to millions of years. While the absolute dose rates might have been low owing to dilution, the total amount of energy liberated in the aquatic environment would be significant and potentially concentrated in biological targets.
Natural fission reactors would clearly be environmentally detrimental. That said, the abundance of uraninite in sediments surrounding oxygen oases cannot have been high enough for natural fission reactors to provide a globally distributed negative feedback on early oxygenic photosynthesic organisms (i.e., at first sight it seems logical that oxygenic photoautotrophs would simply proliferate distal to the natural reactors). This may have been the case. However, there are two plausible scenarios in which natural reactors might have provided a significant negative feedback on the proliferation of oxygenic photoautotrophs: (1) if oxygenic photoautotrophs could only survive under unusual conditions; or (2) if oxygenic photosynthesis evolved in response to environment changes caused by substantial detrital uraninite accumulations.
If the first oxygenic photoautotrophs evolved in unusual environments, they might have been unable to survive away from these specialized environments and hence unable to migrate away if natural fission reactors formed. For example, the availability of fixed nitrogen in the Archean was intimately tied to the biological reduction of N2 by the nitrogenase enzyme. Biological N2-fixation has a high energy requirement (~16 ATPs per N atom), which is met by coupling the process to the oxidation of organic matter to supply chemical energy (Falkowski and Godfrey, 2008). In Archean oxygen oases, an increased supply of organic matter due to the efficiency of oxygenic photosynthesis and the increased energy yield of aerobic respiration likely elevated local rates of nitrogen fixation and relaxed nitrogen limitation of primary production. Increased nutrient availability in the vicinity of natural reactors may have tied early oxygenic photosynthesizers to these environments despite the potential for increased rates of DNA mutagenesis. The second scenario assumes that the oxidizing microenvironments required to provide the selective pressure for oxygenic photoautotrophs to evolve was produced by close proximity to significant detrital accumulations. Radiolysis of water could occur due to non-critical 235U fission in detrital uraninite deposits producing oxidants. If local oxidation of the surface environment in this way provided the selective pressure for the evolution of oxygenic photoautotrophs, then, by necessity, there was abundant uraninite available in these locations and critical fission reactors could readily have formed. In either of these scenarios, it is possible, although not certain, that natural reactors could have provided a (additional) negative feedback to suppress the proliferation of the earliest photoautotrophs and delay the oxidation of Earths surface.
The formation of natural reactors during the late Archean in response to the formation of local oxygen oases is expected based simply on the high abundance of 235U at this time and the redox sensitivity of U solubility. We conclude by noting that irrespective of whether the formation of these natural reactors had any significant biocidal impacts, the geological, geochemical, and biological impacts of natural reactors during this time period deserve further investigation. Because near-surface natural reactors will generally have been eroded away rapidly after formation, searching for them in the geological record is unlikely to prove fruitful. The hypothesis that natural reactors were common in the Archean can be tested, however, by determining the concentration of stable fissogenic nuclides in Archean sediments (or 235U depletion), although this will require very high precision measurements. The impact of near-surface natural reactors on the Archean biosphere is more difficult to determine. Investigation of the evolution of radiation tolerance in some bacteria (e.g., Deinococcus radiodurans and members of the cyanobacteria), for which there is no other obvious terrestrial selective pressure (Sghaier et al., 2007), may prove fruitful.
ACKNOWLEDGMENTS
A highly stimulating review by Zachary Adam contributed significantly to our thinking and is gratefully acknowledged. Paul Falkowski and Gawen Jenkin are also thanked for comments on an early version of the manuscript, and Dante Canil is thanked for discussion. The authors contributed equally to this work and are listed alphabetically.
REFERENCES CITED
- Adam, Z., 2007, Actinides and lifes origins: Astrobiology, v. 7, p. 852872, doi: 10.1089/ast.2006.0066.
- Anbar, A.D., Duan, Y., Lyons, T.W., Arnold, G.L., Kendall, B., Creaser, R.A., Kaufman, A.J., Gordon, G.W., Scott, C., Garvin, J., and Buick, R., 2007, A whiff of oxygen before the Great Oxidation Event?: Science, v. 317, p. 19031906, doi: 10.1126/science.1140325.
- Bekker, A., Holland, H.D., Wang, P.L., Rumble, D., Sytein, H.J., Hannah, J.L., Coetzee, L.L., and Beukes, N.J., 2004, Dating the rise of atmospheric oxygen: Nature, v. 427, p. 117120, doi: 10.1038/nature02260.
- Blankenship, R.E., and Hartman, H., 1998, The origin and evolution of oxygenic photosynthesis: Trends in Biochemical Sciences, v. 23, p. 9497, doi: 10.1016/S0968-0004(98)01186-4.
- Brocks, J.J., Logan, G.A., Buick, R., and Summons, R.E., 1999, Archean molecular fossils and the early rise of eukaryotes: Science, v. 285, p. 10331036.
- Buick, R., 2008, When did oxygenic photosynthesis evolve?: Philosophical Transactions of the Royal Society of London: Series B, v. 363, p. 27312743.
- Canfield, D.E., 2005, The early history of atmospheric oxygen: Homage to Robert A. Garrels: Annual Review of Earth and Planetary Sciences, v. 33, p. 136, doi: 10.1146/annurev.earth.33.092203.122711.
- Canil, D., 1997, Vanadium partitioning and the oxidation state of Archean komatiite magmas: Nature, v. 389, p. 842845, doi: 10.1038/39860.
- Catling, D.C., and Claire, M.W., 2005, How Earths atmosphere evolved to an oxic state: A status report: Earth and Planetary Science Letters, v. 237, p. 120, doi: 10.1016/j.epsl.2005.06.013.
- Catling, D.C., Zahnle, K.J., and McKay, C.P., 2001, Biogenic methane, hydrogen escape, and the irreversible oxidation of early Earth: Science, v. 293, p. 839843, doi: 10.1126/science.1061976.
- DOE (Department of Energy), 1993, Fundamentals Handbook: Nuclear physics and reactor theory, DOE-HDBK-1019/2-93, U.S. Department of Energy, Washington, D.C., v. 2, module 3, 58 p.
- Domagal-Goldman, S.D., Kasting, J.F., Johnston, D.T., and Farquhar, J., 2008, Organic haze, glaciations and multiple sulfur isotopes in the Mid-Archean Era: Earth and Planetary Science Letters, v. 269, p. 2940.
- Draganic, I.G., Graganic, Z.D., and Altiparmakov, D., 1983, Natural nuclear reactors and ionising radiation in the Precambrian: Precambrian Research, v. 20, p. 283298, doi: 10.1016/0301-9268(83)90077-3.
- Falkowski, P.G., and Godfrey, L.V., 2008, Electrons, life and the evolution of Earths oxygen cycle: Philosophical Transactions of the Royal Society of LondonA, v. 363, p. 27052716, doi: 10.1098/rstb.2008.0054.
- Farquhar, J., Bao, H., and Theimens, M., 2000, Atmospheric influence of Earths earliest sulphur cycle: Science, v. 289, p. 756758, doi: 10.1126/science.289.5480.756.
- Fermi, E., 1947, Elementary theory of the chain-reacting pile: Science, v. 105, p. 2732, doi: 10.1126/science.105.2715.27.
- Gaucher, E.A., Govindarajan, S., and Ganesh, O.K., 2008, Palaeotemperature trend for Precambrian life inferred from resurrected proteins: Nature, v. 451, p. 704707, doi: 10.1038/nature06510.
- Gauthier-Lafaye, F., Holliger, P., and Blanc, P.L., 1996, Natural fission reactors in the Franceville basin, Gabon: A review of the conditions and results of a critical event in a geological system: Geochimica et Cosmochimica Acta, v. 60, p. 48314852, doi: 10.1016/S0016-7037(96)00245-1.
- Goldblatt, C., Lenton, T.M., and Watson, A.J., 2006, Bistability of atmospheric oxygen and the Great Oxidation: Nature, v. 443, p. 683686, doi: 10.1038/nature05169.
- Gauthier-Lafaye, F., and Weber, F., 2003, Natural fission reactors: Time constraints for occurrence, and their relation to uranium and manganese deposits and to the evolution of the atmosphere: Precambrian Research, v. 120, p. 81100, doi: 10.1016/S0301-9268(02)00163-8.
- Hidaka, H., and Holliger, P., 1998, Geochemical and neutronic characteristics of the natural fossil fission reactors at Oklo and Bangombé, Gabon: Geochimica et Cosmochimica Acta, v. 62, p. 89108, doi: 10.1016/S0016-7037(97)00319-0.
- Hidaka, H., Holliger, P., and Gauthier-Lafaye, F., 1999, Tc/Ru fractionation in the Oklo and Bangombé natural fission reactors, Gabon: Chemical Geology, v. 155, p. 323333, doi: 10.1016/S0009-2541(98)00173-9.
- Holland, H.D., 2002, Volcanic gases, black smokers, and the Great Oxidation Event: Geochimica et Cosmochimica Acta, v. 66, p. 38113826, doi: 10.1016/S0016-7037(02)00950-X.
- Holland, H.D., 2006, The oxygenation of the atmosphere and oceans: Philosophical Transactions of the Royal Society of London: Series B, v. 361, p. 903915, doi: 10.1098/rstb.2006.1838.
- Karam, P.A., and Leslie, S.A., 1999, Calculations of background beta and gamma radiation levels over geologic time: Health Physics, v. 77, p. 662667, doi: 10.1097/00004032-199912000-00010.
- Karam, P.A., Leslie, S.A., and Anbar, A., 2001, The effects of changing atmospheric oxygen concentrations and background radiation levels on radiogenic DNA damage rates: Health Physics, v. 81, p. 545553, doi: 10.1097/00004032-200111000-00009.
- Kasting, J.F., Holland, H.D., and Pinto, J.P., 1985, Oxidant abundances in rainwater and the evolution of atmospheric oxygen: Journal of Geophysical Research, v. 90, p. 10,49710,510, doi: 10.1029/JD090iD06p10497.
- Kasting, J.F., Eggler, D.H., and Raeburn, S.P., 1993, Mantle redox and the oxidation state of the Archean atmosphere: The Journal of Geology, v. 101, p. 245257.
- Kimberley, M.M., 1978, Origin of stratiform uranium deposits in sandstone, conglomerate, and pyroclastic rock, in Kimberley, M.M., ed., Uranium deposits, their mineralogy and origin: Toronto, University of Toronto Press, p. 339381.
- Kump, L.R., and Barley, M.E., 2007, Increased subaerial volcanism and the rise of atmospheric oxygen 2.5 billion years ago: Nature, v. 448, p. 10331036, doi: 10.1038/nature06058.
- Kump, L.R., Kasting, J.F., and Barley, M.E., 2001, Rise of atmospheric oxygen and the upside-down Archean mantle: Geochemistry Geophysics Geosystems, v. 2, doi: 10.1029/2000GC000114.
- Kuroda, P.K., 1956, On the physical stability of the uranium minerals: The Journal of Chemical Physics, v. 25, p. 781782, doi: 10.1063/1.1743058.
- Lasaga, A.C., 1998, Kinetic theory in the earth science: Princeton, New Jersey, Princeton University Press, 811 p.
- Li, Z.-X.A., and Lee, C.-T. A., 2004, The constancy of upper mantle fO2 through time inferred from V/Sc ratios in basalts: Earth and Planetary Science Letters, v. 228, p. 483493.
- Lowe, D.R., and Tice, M.M., 2007, Tectonic controls on atmospheric, climatic, and biological evolution 3.52.4 Ga: Precambrian Research, v. 158, p. 177197, doi: 10.1016/j.precamres.2007.04.008.
- Mathieu, R., Zetterstrom, L., Cuney, M., Gauthier-Lafaye, F., and Hidaka, H., 2001, Alteration of monazite and zircon and lead migration as geochemical tracers of fluid paleocirculations around the OkloOkelobondo and Bangombé natural nuclear reaction zones Franceville basin, Gabon: Chemical Geology, v. 171, p. 147171, doi: 10.1016/S0009-2541(00)00245-X.
- McKay, C.P., and Hartman, H., 1991, Hydrogen-peroxide and the evolution of oxygenic photosynthesis: Origins of Life and Evolution of the Biosphere, v. 21, p. 157163, doi: 10.1007/BF01809444.
- Meshik, A.P., Hohenberg, C.M., and Pravdivtseva, O.V., 2004, Record of cycling operation of the natural nuclear reactor in the Oklo/Okelobondo area in Gabon: Physical Review Letters, v. 93, p. 182302-1182302-4.
- Nisbet, E.G., Grassineau, N.V., Howe, C.J., Abell, P.I., Regelous, M., and Nisbet, R.E.R., 2007, The age of Rubisco: the evolution of oxygenic photosynthesis: Geobiology, v. 5, p. 311335, doi: 10.1111/j.1472-4669.2007.00127.x.
- Ohmoto, H., Watanabe, Y., Ikemi, H., Poulson, S.P., and Taylor, B.E., 2006, Sulphur isotope evidence for an oxic Archaean atmosphere: Nature, v. 442, p. 908911, doi: 10.1038/nature05044.
- Ono, S., 2001, Detrital uraninite and the early Earths atmosphere: SIMS analyses of uraninite in the Elliot lake district and the dissolution kinetics of natural uraninite [Ph.D. thesis]: Pennsylvania State University, 209 p.
- Petrov, Y.V., Nazarov, A.I., Onegin, M.S., Petrov, V.Y., and Sakhnovsky, E.G., 2006, Natural nuclear reactors at Oklo and variation of fundamental constants: Computation of neutronics of a fresh core: Physical Review C, v. c74, p. 064610-1064610-17.
- Rasmussen, B., Fletcher, I.R., Brocks, J.J., and Kilburn, M.R., 2008, Reassessing the first appearance of eukaryotes and cyanobacteria: Nature, v. 455, p. 11011105, doi: 10.1038/nature07381.
- Rye, R., and Holland, H.D., 1998, Paleosols and the evolution of atmospheric oxygen: A critical review: American Journal of Science, v. 298, p. 621672.
- Rosing, M.T., and Frei, R., 2004, U-rich seafloor sediments from GreenlandIndications of >3700 Ma oxygenic photosynthesis: Earth and Planetary Science Letters, v. 217, p. 237244, doi: 10.1016/S0012-821X(03)00609-5.
- Schopf, J.W., 1993, Microfossils of the Early Archean Apex ChertNew evidence of the antiquity of life: Science, v. 260, p. 640646.
- Sghaier, H., Narumi, I., Satoh, K., Ohba, H., and Mitomo, H., 2007, Problems with the current deinococcal hypothesis: An alternative theory: Theory in Biosciences, v. 126, p. 4345, doi: 10.1007/s12064-007-0004-x.
- Theis, N.J., 1978, Mineralogy and setting of Elliot lake deposits, in Kimberley, M.M., ed., Uranium deposits, their mineralogy and origin: Toronto, University of Toronto Press, p. 331338.
- Tian, F., Toon, O.B., Pavlov, A.A., and Sterck, H.D., 2005, A hydrogen-rich early Earth atmosphere: Science, v. 308, p. 10141017, doi: 10.1126/science.1106983.
- Wilkinson, B.H., and Kesler, S.E., 2007, Tectonism and exhumation in convergent margin orogens: Insights from ore deposits: The Journal of Geology, v. 115, p. 611627.